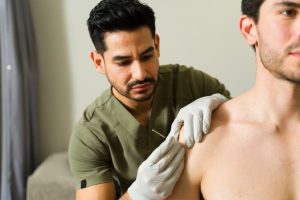
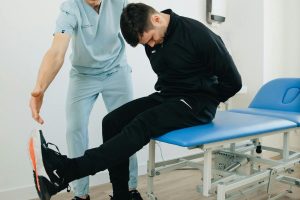
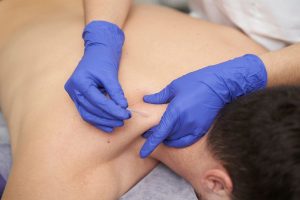
Over the last 30 years, we’ve worked with many Ehlers-Danlos syndrome patients who benefitted directly from physical therapy.
In this article, we’ll discuss exactly what Ehlers-Danlos syndrome is, how it is diagnosed, and what treatment options are available.
What is EDS?
The Ehlers-Danlos syndromes are a group of connective tissue disorders that can be inherited and are varied both in how affect the body and in their genetic causes. They are generally characterized by joint hypermobility (joints that stretch further than normal), skin hyperextensibility (skin that can be stretched further than normal), and tissue fragility.
The Ehlers-Danlos syndromes (EDS) are currently classified into thirteen subtypes. Each EDS subtype has a set of clinical criteria that help guide diagnosis; a patient’s physical signs and symptoms will be matched up to the major and minor criteria to identify the subtype that is the most complete fit. There is substantial symptom overlap between the EDS subtypes and the other connective tissue disorders including hypermobility spectrum disorders, as well as a lot of variability, so a definitive diagnosis for all the EDS subtypes when the gene mutation is known—all but hypermobile EDS (hEDS)—also calls for confirmation by testing to identify the responsible variant for the gene affected in each subtype.
For those who meet the minimal clinical requirements for an EDS subtype—but who have no access to molecular confirmation; or whose genetic testing shows one (or more) gene variants of uncertain significance in the genes identified for one of the EDS subtypes; or in whom no causative variants are identified in any of the EDS-subtype-specific genes—a “provisional clinical diagnosis” of an EDS subtype can be made. These patients should be followed clinically, but alternative diagnoses and expanded molecular testing should be considered.
Please remember that an individual’s experience with an EDS is their own, and may not necessarily be the same as another person’s experience. Diagnostic criteria are meant solely to distinguish an EDS from other connective tissue disorders, and there are many more possible symptoms for each EDS than there are criteria.
What are the symptoms of Ehlers-Danlos syndromes?
Clinical manifestations of an Ehlers-Danlos syndrome are most often joint and skin related and may include:
Joints
Joint hypermobility; loose/unstable joints which are prone to frequent dislocations and/or subluxations; joint pain; hyperextensible joints (they move beyond the joint’s normal range); early onset of osteoarthritis.
Skin
Soft velvety-like skin; variable skin hyper-extensibility; fragile skin that tears or bruises easily (bruising may be severe); severe scarring; slow and poor wound healing; development of molluscoid pseudo tumors (fleshy lesions associated with scars over pressure areas).
Miscellaneous/Less Common
Chronic, early onset, debilitating musculoskeletal pain (usually associated with the Hypermobility Type); arterial/intestinal/uterine fragility or rupture (usually associated with the Vascular Type); scoliosis at birth and scleral fragility (associated with the Kyphoscoliosis Type); poor muscle tone (associated with the Arthrochalasia Type); mitral valve prolapse; and gum disease.
Each type of Ehlers-Danlos syndrome is defined as a distinct problem in connective tissue. Connective tissue is what the body uses to provide strength and elasticity; normal connective tissue holds strong proteins that allow tissue to be stretched but not beyond its limit, and then safely return that tissue to normal. Connective tissue is found throughout the body, and Ehlers-Danlos syndromes are structural problems. An analogy: If one builds a house with faulty materials, say half the necessary wood or with soft aluminum nails, it is certain there will be problems. Some problems are more likely to show up than others, but because those materials were used everywhere and are not necessarily visible, one can be surprised by where a problem shows up or how serious it is.
It is much the same thing with an Ehlers-Danlos Syndrome and connective tissue.
The connective tissue a person with EDS is built with is not structured the way it should be. With a badly-constructed or processed connective tissue, some or all of the tissue in the EDS-affected body can be pulled beyond normal limits which causes damage. Connective tissue can be found almost anywhere, in skin, muscles, tendons and ligaments, blood vessels, organs, gums, eyes, and so on.
The problems resulting from one’s body being built out of a protein that behaves unreliably can be widespread and in a wide range of severity. It shows up in places that seem unrelated until the underlying connection to an Ehlers-Danlos syndrome is recognized.
What are the types of Ehlers-Danlos syndrome?
There are thirteen defined types of Ehlers-Danlos syndrome, as well as a number of mutations identified as Ehlers-Danlos syndrome that fall outside the current system. The major types of Ehlers-Danlos syndrome are classified according to the signs and symptoms that are manifested. Each type of Ehlers-Danlos syndrome is a distinct disorder that “runs true” in a family. An individual with Vascular Ehlers-Danlos syndrome will not have a child with Classical Ehlers-Danlos syndrome. Learn more about the different types of Ehlers-Danlos Syndrome.
How is an Ehlers-Danlos Syndrome diagnosed?
If you think you might have one of the Ehlers-Danlos syndromes (EDS) or hypermobility spectrum disorders (HSD), and particularly if someone in your immediate family has been diagnosed, ask your doctor if a diagnosis fits your symptoms. If they choose to, any doctor who can diagnosis a disease is able to diagnose EDS/HSD; but most likely you’ll be given a referral to a geneticist, because EDS are genetic disorders and geneticists are most adept at distinguishing between those diseases, as well as in doing any testing necessary to differentiate EDS/HSD from the more than 200 other heritable connective tissue disorders.
A diagnosis is important because, although EDS/HSD are not curable, they are treatable. Knowing the type of EDS/HSD gives you and your medical team some idea of where problems might come from and why they’re happening. When eventually there is a cure, you’ll know to use it. And as more of us are diagnosed, EDS/HSD gain the attention all of us need, increasing the likelihood of expanded research that might lead to finding that cure.
Your path to an EDS/HSD diagnosis starts with an examination. There may be physical testing: using the Beighton Scale to assess how mobile your joints are, a search for abnormal scarring and testing your skin to determine what it feels like and how much it stretches, as well as any additional tests your particular doctor feels are needed. There’s likely to be a look into your medical history to look for conditions and problems associated with EDS/HSD, and a discussion of your family to help determine if an EDS/HSD was inherited.
Diagnosis of an EDS subtype comes by finding the one that most matches your symptoms. There are clinical criteria that help guide diagnosis; your signs and symptoms will be matched up to the major and minor criteria to identify the subtype that is the most complete fit. There is substantial symptom overlap between the EDS subtypes and the other connective tissue disorders including HSD, as well as a lot of variability between them. So a definitive diagnosis for all the EDS subtypes—except for hypermobile EDS (hEDS)—also calls for confirmation by testing to identify the responsible variant for the gene affected in each subtype. These molecular testing results also provide the basis for genetic counseling for our families, guidance on treatment options for ourselves, and help in reaching research goals.
The genetic basis for hypermobile EDS is still unknown, so an hEDS (or HSD) diagnosis rests on the criteria and what your doctor finds during your examination. The hEDS criteria also established serious consideration of joint hypermobility with all related symptoms and conditions, with hEDS at one end of the spectrum. HSD can be no less consequential than hEDS, either to your health or concern for treatment.
You can find the diagnostic criteria for the 13 subtypes of EDS by clicking here.
How prevalent are Ehlers-Danlos Syndromes?
At this time, research statistics of the Ehlers-Danlos syndromes show the total prevalence as 1 in 2,500 to 1 in 5,000 people. Recent clinical experience suggests that Ehlers-Danlos syndrome may be more common. The conditions are known to affect both males and females of all racial and ethnic backgrounds.
How are Ehlers-Danlos syndromes inherited?
The two known inheritance patterns for the Ehlers-Danlos syndromes include autosomal dominant and autosomal recessive. Regardless of the inheritance pattern, we have no choice in which genes we pass on to our children.
What is the prognosis of someone with an Ehlers-Danlos Syndrome?
The prognosis depends on the type of Ehlers-Danlos syndrome and the individual. Life expectancy can be shortened for those with the Vascular Ehlers-Danlos syndrome due to the possibility of organ and vessel rupture. Life expectancy is usually not affected in the other types. There can be a wide or narrow range of severity within a family, but each person’s case of Ehlers-Danlos syndrome will be unique. While there is no cure for the Ehlers-Danlos syndromes, there is treatment for symptoms, and there are preventative measures that are helpful for most.
What can I do now?
The Ehlers-Danlos Society members are sharing information online and learning from each other in ways that were impossible not very long ago. Visit The Ehlers-Danlos Society’s Facebook page or The Ehlers-Danlos Society Inspire Online Community.
How to Treat EDS:
- Strength and stability exercises
- Activity modification
- Pain management/control methods
- Taping and bracing
- Dry Needling
- Emotional support and counselling
- Medication